Isolated kidney transplantation under Lumasiran therapy in primary hyperoxaluria type 1: A case report
Diletta Torres1, Donatella Simone1, Simona Simone2, Michele Rossini2, Fausta Piancone2, Mario Giordano1, Pasquale Di Tonno3, Loreto Gesualdo2.
1Department of Pediatrics, Pediatric Nephrology and Dialysis Unit Giovanni XXIII Hospital , Bari, Italy; 2Department of Emergency and Organ Transplantation, Nephrology Dialysis and Transplantation Unit, Bari, Italy; 3Department of Emergency and Organ Transplantation, Urology, Andrology and Kidney Transplantation Unit, Bari, Italy
Background: Primary hyperoxaluria type 1 (PH1), is a rare autosomal recessive genetic disorder due to a mutation in the AGXT gene, encoding for alanine-glyoxylate aminotransferase (AGT). When AGT activity is absent or deficient plasma oxalate increase leading to systemic oxalosis. Symptoms range from nephrocalcinosis to recurrent kidney stones, until chronic kidney disease (CKD) and end stage kidney disease (ESKD).Traditionally, combined liver-kidney transplantation (CLKT) has been recommended. Liver transplantation corrects the metabolic defect in PH1, but it is associated with significant morbidity and mortality. Recently, novel therapeutic strategies have emerged, as Lumasiran, an RNA interference. Lumasiran, from November 2020, is the first and only approved drug for PH1. Isolated kidney transplantation (iKT) in combination with Lumasiran is now a viable option. However, whether this therapeutic strategy can fully replace CLKT remains uncertain. Single KT combined with Lumasiran therapy is costly and not universally accessible, in this case CLKT remains the first-line therapeutic option. The literature increasingly supports the use of iKT in combination with Lumasiran. This is the first reported case in Italy of a pediatric patient evaluated for KT alone from a living donor, while receiving intensive HD and Lumasiran.
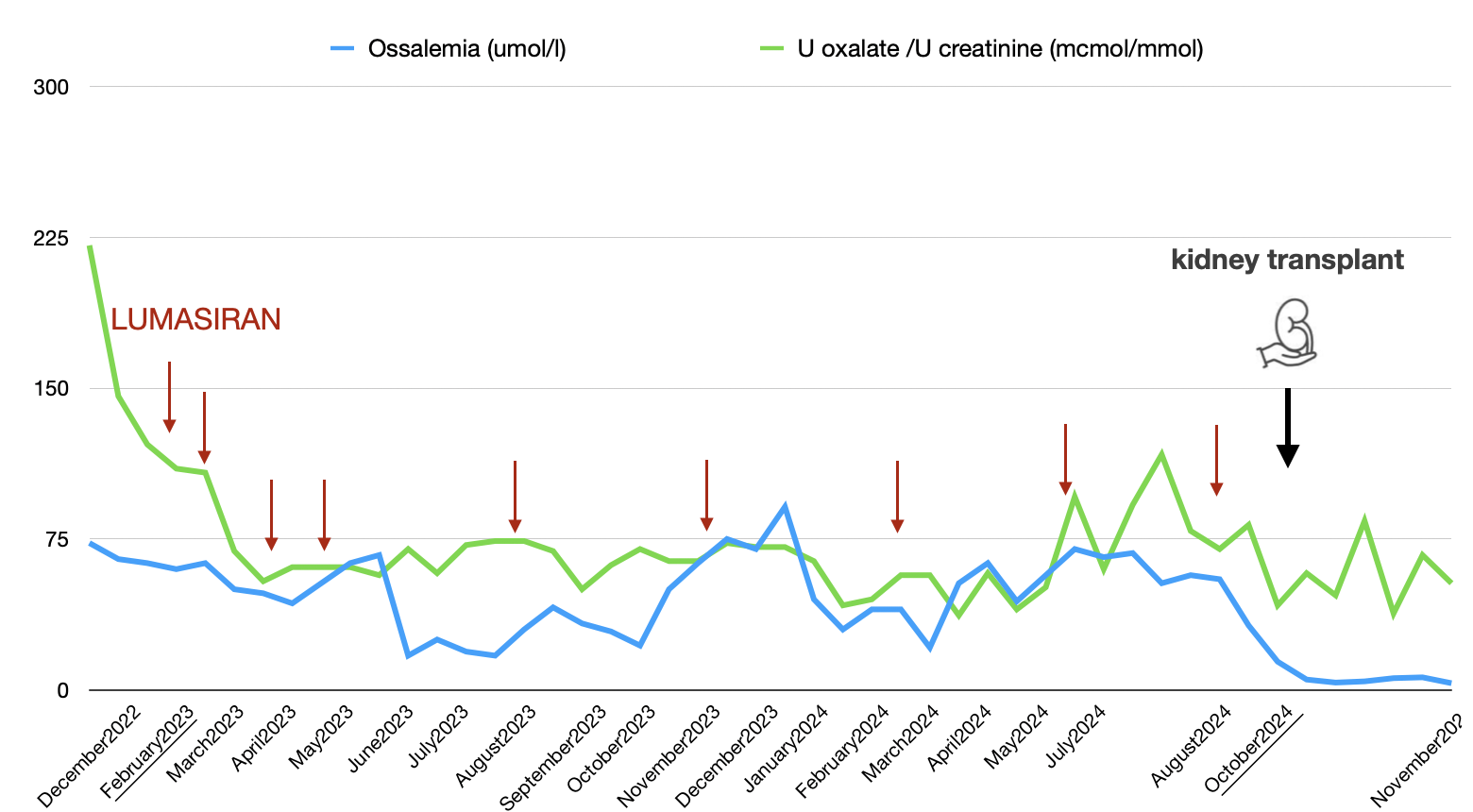
Case report: A 16-year-old Albanian boy diagnosed with PH1 at the age of 5 (point mutation C.508G>A in exon 4 of the AGXT gene), came in our ward to begin iHD. Creatinine was 5.52 mg/dL, Schwartz eGFR 13 ml/min. Urinary oxalate levels (R UOxal/UCrea 221) and oxalemia (300 µmol/L) were high. HD was initiated at a frequency of four sessions per week, alongside therapy with Lumasiran at a dose of 3 mg/kg. Oxalemia and oxaluria levels significantly decreased, reaching values considered safe for a iKT. The patient’s family declined the CLKT program and after six months he underwent a living donor iKT (mother). Lumasiran infusions continued every three months. In the immediate postoperative period, daily HD sessions were performed from day 1 to 7, and at day 9 thereafter.
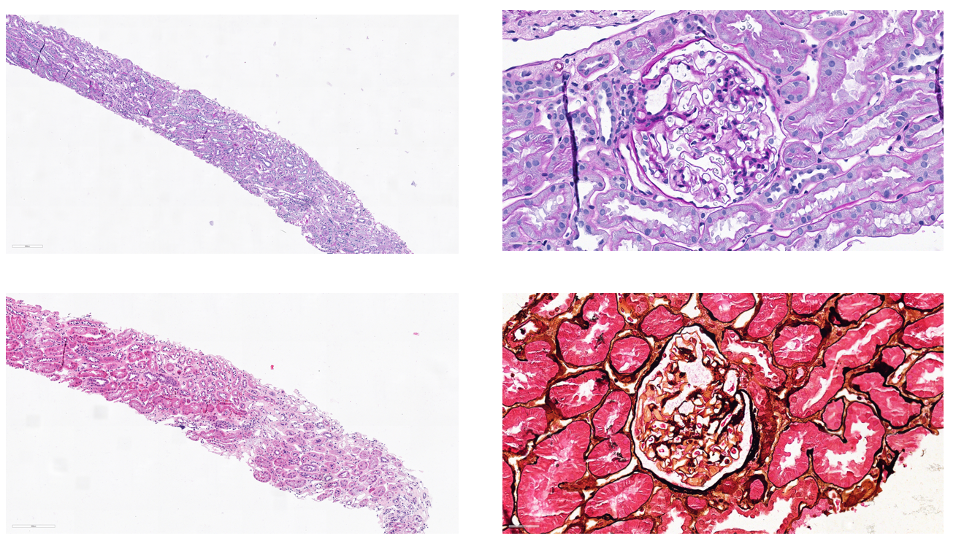
Results: Plasma and urinary oxalates were monitored (Figure 1). The patient was discharged from the KT unit after 16 days, with a serum creatinine of 1.3 mg/dL, BUN of 47 mg/dL, and 24-hour proteinuria of 0.2 g/day. One month later, a protocol kidney biopsy revealed normal glomeruli, tubules, interstitium, and vessels, with a single small deposit of calcium oxalate in a tubular lumen (Image 1).
Conclusions: Our case suggests that iKT combined with Lumasiran therapy may be a viable option for pediatric patients with ESKD due to PH1. However, longer follow-up is necessary to assess the long-term effects of this therapy on the transplanted kidney. Further evidence in the pediatric population is needed to establish new therapeutic recommendations.
Several authors of this publication are members of the European Reference Network for Rare Kidney Diseases (ERKNet).
[1] kidney transplant
[2] lumasiran
[3] PH1
[4] AGXT